Overview
We want to calculate the power to detect an association a genetic marker and a quantitative phenotype.
- Write functions to simulate genotype and phenotype under an additive genetic model.
- Simulate genotype and phenotype using the simulator under the null and alternative.
- Select the relevant statistic and perform the hypothesis test.
- Calculate the power of the test.
As a concrete example, we pre-specify the model for continuous trait \(Y\) and genotype \(X\) as
\[Y = \beta X + \epsilon, ~~~~ \text{with } \epsilon \sim N(0, \sigma_\epsilon^2)\]
where we assume
- genotype of the locus follows Hardy-Weinberg equilibrium with a pre-specified minor allele frequency.
- trait is normalized to variance 1 and the proportion of variation explained by the SNP is \(r^2\).
The goal is to calculate the power with the significance level (\(\alpha\)), effect size (in the form of \(r^2\)), and sample size (num_individuals).
Note that the proportion of variation explained, minor allele frequency, effect size (\(\beta\)) are related under the model above and you will be/have already been asked to work it out in assignment.
Phenotype-genotype simulator
To simulate genotype, we assume the locus is bialleilic and each individual is diploid. So that \(X \sim \text{Binomial}(2, f)\) with \(f\) as minor allele frequency (here we encode minor allele as 1 and major allele as 0).
Given genotype, to simulate phenotype, we need to know \(\beta\) and \(\sigma_\epsilon^2\). So, we first calculate the effect size and the variance of the noise term from the proportion of variation explained, variance of trait, and minor allele frequency of the SNP.
simulate_genotype = function(maf, num_individuals, num_replicates) {
# maf: minor allele frequency
# num_individuals: the number of individuals in each replicates
# num_replicates: the number of replicates
# it returns a matrix with num_individuals rows and num_replicates columns
genotype = matrix(
rbinom(num_individuals * num_replicates, 2, maf),
nrow = num_individuals,
ncol = num_replicates
)
return(genotype)
}
simulate_phenotype = function(genotype, beta, sig2epsi) {
# genotype: each column is one replicate
# beta: effect size of the linear model
# sig2epsi: the variance of the noise term
num_individuals = nrow(genotype)
num_replicates = ncol(genotype)
epsilon = matrix(
rnorm(num_individuals * num_replicates, mean = 0, sd = sqrt(sig2epsi)),
nrow = num_individuals,
ncol = num_replicates
)
phenotype = genotype * beta + epsilon
return(phenotype)
}
get_beta_and_sig2epsi = function(r2, sig2Y, maf) {
# r2: proportion of variation explained by SNP
# sig2Y: variance of trait
# maf: minor allele frequency of SNP
# it returns a list of beta and variance of noise term
## effect size is calculated from r2 = beta^2 *2*maf*(1-maf)
beta = sqrt( r2 * sig2Y / (2 * maf * (1 - maf)) )
sig2epsi = sig2Y * (1 - r2)
return(list(beta = beta, sig2epsi = sig2epsi))
}
linear_model_simulator = function(num_replicates, num_individuals, maf, r2, sig2Y) {
# simulate genotype
X = simulate_genotype(maf, num_individuals, num_replicates)
# calculate model parameters
params = get_beta_and_sig2epsi(r2, sig2Y, maf)
# simulate phenotype given genotype and model parameters
Y = simulate_phenotype(X, params$beta, params$sig2epsi)
return(list(Y = Y, X = X))
}Simulate 10,000 Y vectors under null and alternative scenarios
Here we simulate 1000 individuals per replicate and 10000 replicates in total. With parameters: * Minor allele frequency is 0.3. * Proportion of variance explained is 0.01 * Variance of trait is 1.
# specify paramters
nindiv = 1000
nreplicate = 10000
maf = 0.30
r2 = 0.01
sig2Y = 1
# run simulator
## under the alternative
data_alt = linear_model_simulator(nreplicate, nindiv, maf, r2, sig2Y)
dim(data_alt$Y)## [1] 1000 10000dim(data_alt$X)## [1] 1000 10000## under the null
data_null = linear_model_simulator(nreplicate, nindiv, maf, 0, sig2Y)
dim(data_null$Y)## [1] 1000 10000dim(data_null$X)## [1] 1000 10000define functions to run associations and obtain the z-scores
runassoc = function(X,Y)
{
pvec = rep(NA,ncol(X))
bvec = rep(NA,ncol(X))
for(ss in 1:ncol(X))
{
fit = fastlm(X[,ss], Y[,ss])
pvec[ss] = fit$pval
bvec[ss] = fit$betahat
}
list(pvec=pvec, bvec=bvec)
}
p2z = function(b,p)
{
## calculate zscore from p-value and sign of effect size
sign(b) * abs(qnorm(p/2))
}
calcz = function(X,Y)
{
tempo = runassoc(X,Y)
p2z(tempo$bvec,tempo$pvec)
}Load some packages
library(tidyverse)## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.0 ──## ✓ ggplot2 3.3.3 ✓ purrr 0.3.4
## ✓ tibble 3.0.5 ✓ dplyr 1.0.3
## ✓ tidyr 1.1.2 ✓ stringr 1.4.0
## ✓ readr 1.4.0 ✓ forcats 0.5.0## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## x dplyr::filter() masks stats::filter()
## x dplyr::lag() masks stats::lag()fastlm = function(xx,yy)
{
## compute betahat (regression coef) and pvalue with Ftest
## for now it does not take covariates
df1 = 2
df0 = 1
ind = !is.na(xx) & !is.na(yy)
xx = xx[ind]
yy = yy[ind]
n = sum(ind)
xbar = mean(xx)
ybar = mean(yy)
xx = xx - xbar
yy = yy - ybar
SXX = sum( xx^2 )
SYY = sum( yy^2 )
SXY = sum( xx * yy )
betahat = SXY / SXX
RSS1 = sum( ( yy - xx * betahat )^2 )
RSS0 = SYY
fstat = ( ( RSS0 - RSS1 ) / ( df1 - df0 ) ) / ( RSS1 / ( n - df1 ) )
pval = 1 - pf(fstat, df1 = ( df1 - df0 ), df2 = ( n - df1 ))
res = list(betahat = betahat, pval = pval)
return(res)
}Now we can calculate test statistics under the null and alternative scenarios.
Zalt = calcz(data_alt$X, data_alt$Y)
Znull = calcz(data_null$X, data_null$Y)
## create a data frame to use ggplot
pp = tibble(Y = c(Zalt,Znull), type=c(rep("alt",length(Zalt)),rep("null",length(Znull))) ) %>% ggplot(aes(Y,fill=type)) + geom_density(color=NA,alpha=0.6) + theme_bw(base_size = 15)
pp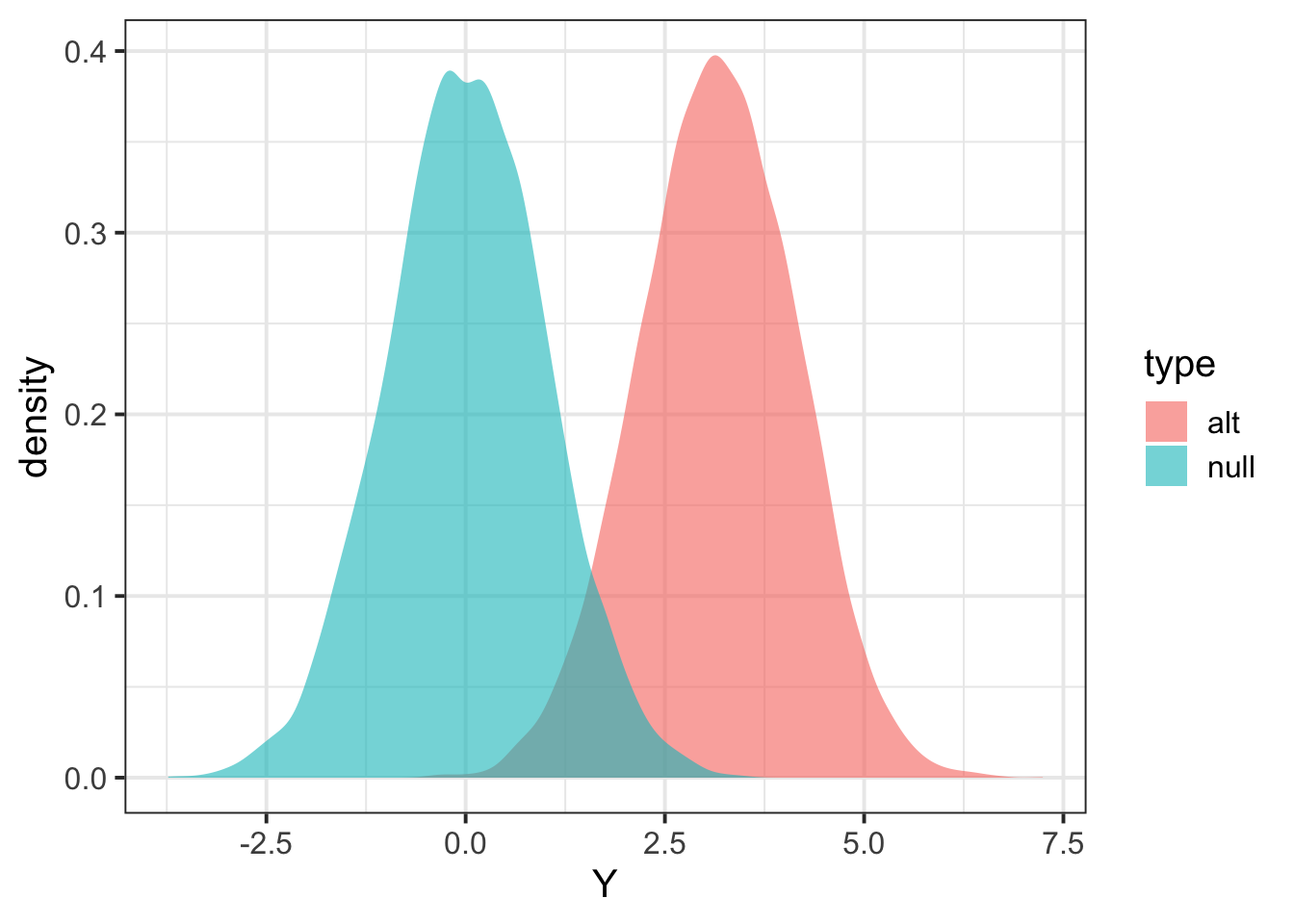
Calculate power
## define significance level (=type I error we are willing to accept)
alpha = 0.01
## find threshold for rejection; we want P(Znull > alpha/2) two-sided
threshold = quantile(Znull, 1 - alpha/2)
print(threshold,2)## 99.5%
## 2.6## add threshold to the Zscores figure
pp + geom_vline(xintercept=threshold,col='dark green',size=1.5)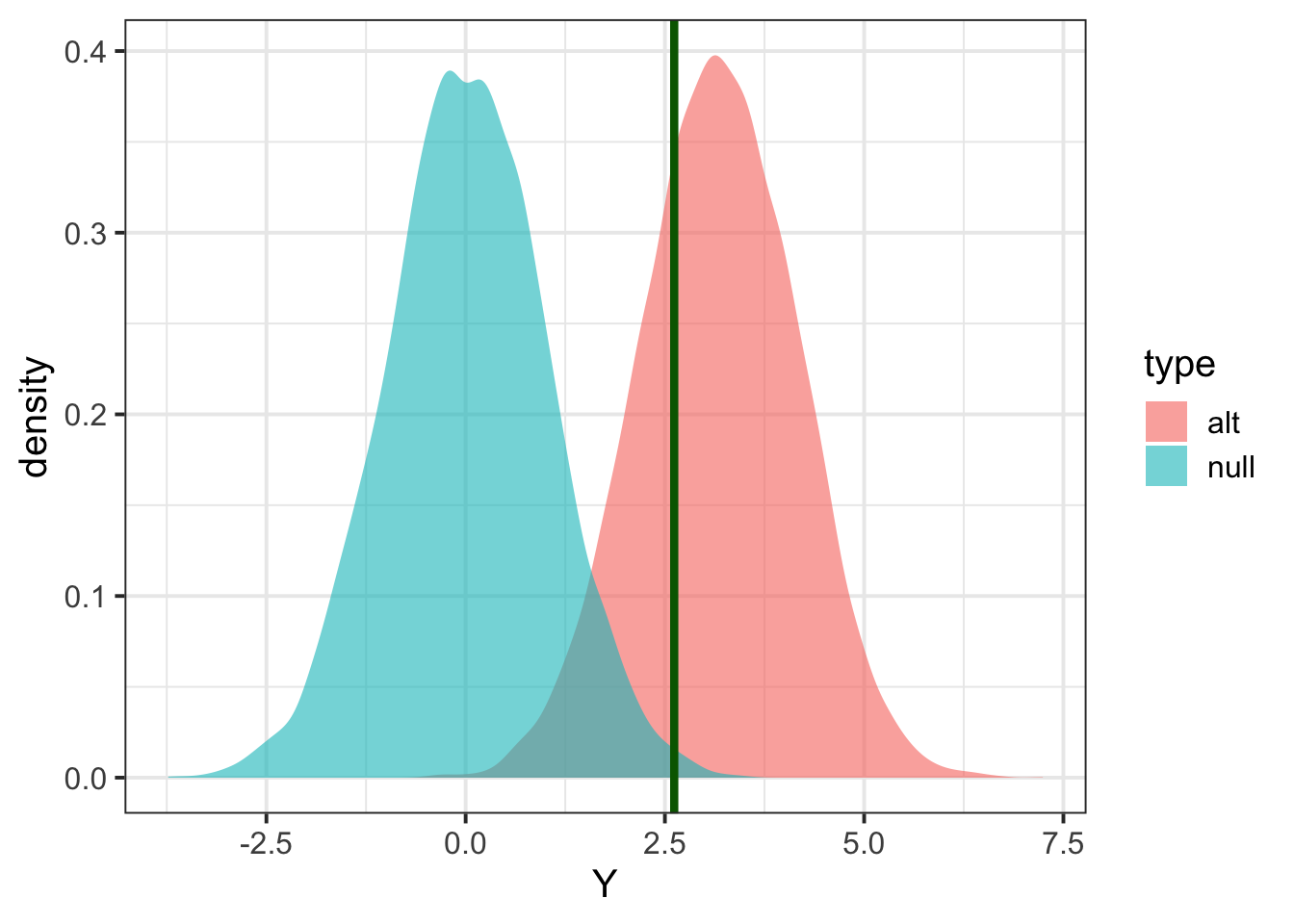
## calculate proportion of Zalt above threshold
power = mean(Zalt > threshold)
print(power)## [1] 0.7106## to be more precise, we should be calculating the proportion of null replicates that are either above or
mean(Zalt > threshold | Zalt < -threshold)## [1] 0.7106## not much different since the probability of Zalt being negative is almost 0check with pwr.r.test function
## install.packages("pwr")
if (!("pwr" %in% installed.packages()[, 1])) {
install.packages("pwr")
}
pwr::pwr.r.test(n = nindiv, r= sqrt(r2), sig.level = alpha)##
## approximate correlation power calculation (arctangh transformation)
##
## n = 1000
## r = 0.1
## sig.level = 0.01
## power = 0.7234392
## alternative = two.sided