Goal
-[ ] download Personal Genome Project data (http://repgp.teamerlich.org/)
-[ ] analyze content, explore structure
-[ ] predict height and other phenotypes with PRS and compare to reported values
library(tidyverse)## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.0 ──## ✓ ggplot2 3.3.3 ✓ purrr 0.3.4
## ✓ tibble 3.1.0 ✓ dplyr 1.0.3
## ✓ tidyr 1.1.2 ✓ stringr 1.4.0
## ✓ readr 1.4.0 ✓ forcats 0.5.0## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## x dplyr::filter() masks stats::filter()
## x dplyr::lag() masks stats::lag()library(RSQLite)
sqlite <- dbDriver("SQLite")
DATA="/Users/haekyungim/Box/LargeFiles/imlab-data/data-Github/web-data/2021-04-21-personal-genomes-project-data/"
WORK=DATADefine environmental variables for convenience
DATA="/Users/haekyungim/Box/LargeFiles/imlab-data/data-Github/web-data/2021-04-21-personal-genomes-project-data/"
WORK=$DATA
PRSice=/Users/haekyungim/bin/PRSice_mac
BIN="/Users/haekyungim/bin"## download sqqtl database with phenotypes?
cd $DATA
wget http://files.teamerlich.org/repgp/repgp-data.sqlite3
wget http://files.teamerlich.org/repgp/repgp.vcf.gz
dbname <- paste0(DATA,"repgp-data.sqlite3") ## add full path if db file not in current directory
## connect to db
db = dbConnect(sqlite,dbname)
## list tables
dbListTables(db)## [1] "ancestry_jkp" "ancestry_pop" "ancestry_supop" "users"dbListFields(db, "users") ## [1] "id" "sample" "height" "weight" "human_id" "file_id" "blood"
## [8] "gender" "race"## convenience query function
query <- function(...) dbGetQuery(db, ...)users = query("select * from users")
names(users)## [1] "id" "sample" "height" "weight" "human_id" "file_id" "blood"
## [8] "gender" "race"users %>% count(race)## race n
## 1 * 68
## 2 American Indian or Alaska Native 2
## 3 Asian 6
## 4 Black or African American 1
## 5 Caucasian (White) 1
## 6 Hispanic or Latino 2
## 7 Hispanic/Latino 3
## 8 White 167users %>% count(gender)## gender n
## 1 * 69
## 2 Female 46
## 3 Male 135users %>% count(blood)## blood n
## 1 * 111
## 2 A- 10
## 3 A+ 41
## 4 AB- 1
## 5 AB+ 4
## 6 B- 4
## 7 B+ 16
## 8 O- 11
## 9 O+ 52users %>% ggplot(aes(height,fill=gender)) + geom_density(alpha=0.6) + ggtitle("Height by gender - Missing gender, *, has bi-modal distr.")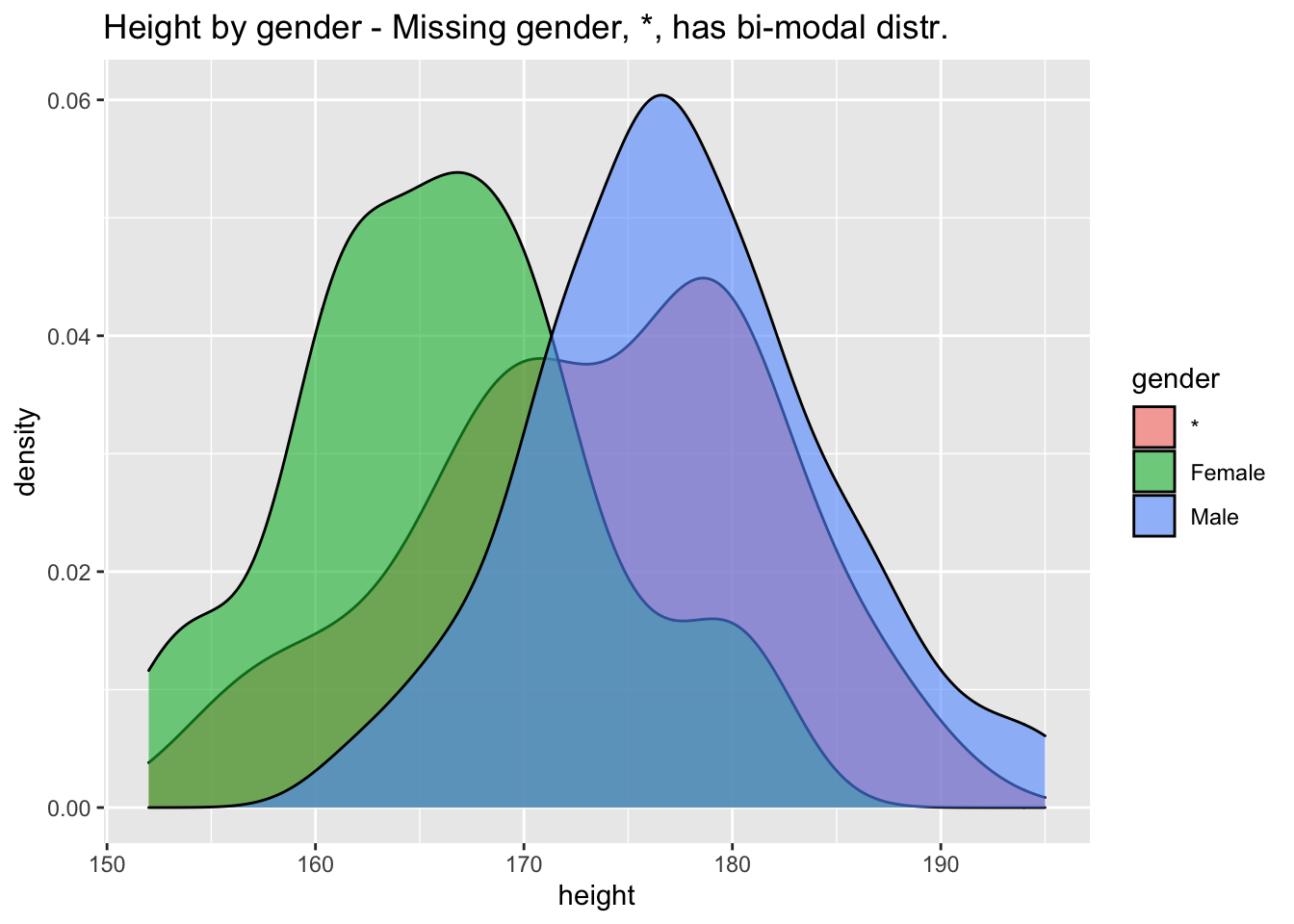
Download imputed genotype files
cd $DATA
for num in $(seq 22);
do
wget http://files.teamerlich.org/repgp/repgp-imputed.chr${num}.vcf.gz
doneCreate plink formatted genotype files. You will need plink2, which you can download from here
## install plink2 from https://www.cog-genomics.org/plink/2.0/
## more plink2 to ~/bin/
## ~/bin/plink2 --vcf repgp-imputed.chr22.vcf.gz --make-bed --out repgp
cd $DATA
for num in $(seq 22);
do
~/bin/plink2 --vcf repgp-imputed.chr${num}.vcf.gz --make-bed --out repgp.chr${num}
doneChoose a trait and download its GWAS summary statistics from this list https://uchicago.box.com/v/test-gtex-gwas-ukb-data
## here I chose sleep duration
cd $DATA
##wget https://uchicago.box.com/s/4gc4u1yr579qjbz8s6d27h8vb0h1yw1q
wget -O ukb_sleep_duration.gz https://uchicago.box.com/shared/static/4gc4u1yr579qjbz8s6d27h8vb0h1yw1q.gz
## height
wget -O ukb_height.gz https://uchicago.box.com/shared/static/2889tf5fx5d5ulll7otg4dltd1vogabh.gzCreate phenotype file
fam = read_tsv(glue::glue("{DATA}/repgp.fam"),col_names = FALSE)##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## X1 = col_double(),
## X2 = col_character(),
## X3 = col_double(),
## X4 = col_double(),
## X5 = col_double(),
## X6 = col_double()
## )names(fam)[1:2] = c("FID","IID")
fam <- fam %>% select(FID, IID) %>% inner_join(users %>% select(sample,height,weight,gender), by=c("IID"="sample"))
write_tsv(fam,file=glue::glue("{DATA}/phenodata.txt"))## use --pheno-col to specify phenotype list
mkdir $WORK/output
Rscript $PRSice/PRSice.R --dir $PRSice \
--prsice $PRSice/PRSice_mac \
--base $WORK/ukb_height.gz \
--target $WORK/repgp.chr# \
--snp variant_id \
--A1 effect_allele \
--A2 non_effect_allele \
--stat effect_size \
--beta \
--pvalue pvalue \
--pheno-file $WORK/phenodata.txt \
--pheno-col height \
--bar-levels 5e-08,5e-07,5e-06,5e-05,5e-04,5e-03,5e-02,5e-01,1 \
--fastscore \
--binary-target F \
--thread 2 \
--out $WORK/output/height_score_allGot the following error. Some SNP IDs are duplicated.
Error: A total of 14 duplicated SNP ID detected out of
6417846 input SNPs! Valid SNP ID (post --extract /
--exclude, non-duplicated SNPs) stored at
/Users/haekyungim/Box/LargeFiles/imlab-data/data-Github/web-data/2021-04-21-personal-genomes-project-data//output/height_score_all.valid.
You can avoid this error by using --extract
/Users/haekyungim/Box/LargeFiles/imlab-data/data-Github/web-data/2021-04-21-personal-genomes-project-data//output/height_score_all.valid Try again including only the SNP list created by the previous run of PRSice, height_score_all.valid A1 is the effect allele in PRSice, similar to Plink’s default. Other software use different conventions so this sign is something that need to be checked carefully.
Rscript $PRSice/PRSice.R --dir $PRSice \
--prsice $PRSice/PRSice_mac \
--base $WORK/ukb_height.gz \
--target $WORK/repgp.chr# \
--snp variant_id \
--A1 effect_allele \
--A2 non_effect_allele \
--stat effect_size \
--beta \
--pvalue pvalue \
--pheno-file $WORK/phenodata.txt \
--pheno-col height \
--bar-levels 5e-08,5e-07,5e-06,5e-05,5e-04,5e-03,5e-02,5e-01,1 \
--fastscore \
--binary-target F \
--thread 2 \
--extract $WORK/output/height_score_all.valid \
--out $WORK/output/height_score_allThis time it worked.
 This figure shows the correlation (squared) between prediction and actual values provided in the phenodata.txt file. In this dataset, using SNPs with p-values up to 0.005 yielded the most significant correlation. Next, we will plot the predicted and actual values to get a better sense of how well PRS is predicting.
This figure shows the correlation (squared) between prediction and actual values provided in the phenodata.txt file. In this dataset, using SNPs with p-values up to 0.005 yielded the most significant correlation. Next, we will plot the predicted and actual values to get a better sense of how well PRS is predicting.
Plot predicted vs actual height
## we already have the phenotype data in the fam variable
predicted_height = read_delim(glue::glue("{DATA}/output/height_score_all.best"),delim=" ")##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## FID = col_double(),
## IID = col_character(),
## In_Regression = col_character(),
## PRS = col_double()
## )combined <- predicted_height %>% inner_join(fam,by=c("IID"="IID"))
combined %>% ggplot(aes(PRS,height)) + geom_point()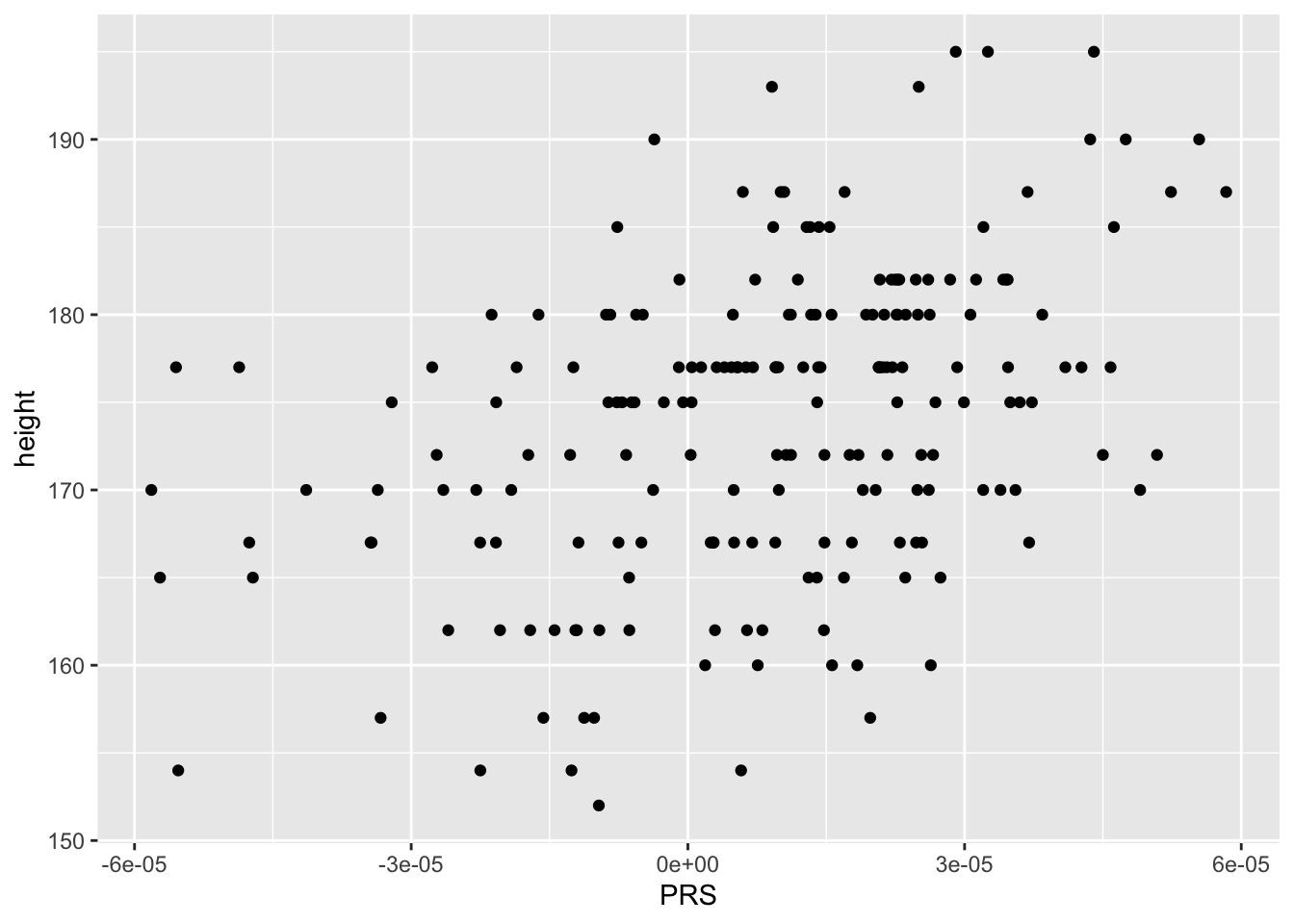
summary(lm(height ~ PRS,data=combined)) %>% coef() ## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 172.6716 6.075067e-01 284.230021 1.916175e-259
## PRS 159197.1399 2.478220e+04 6.423849 9.758137e-10