Goals and Introduction
In this lab we are going to use the PRSice software to take height GWAS results and predict height of the Personal Genome Project individuals using their genotype data.
Goals
- Download data from box
- Download PRSice
- Run exploratory analysis of data
- Run PRSice
- Plot observed versus predicted for height
Setup
R Libraries
library(tidyverse)## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──## ✓ ggplot2 3.3.5 ✓ purrr 0.3.4
## ✓ tibble 3.1.6 ✓ dplyr 1.0.8
## ✓ tidyr 1.2.0 ✓ stringr 1.4.0
## ✓ readr 2.1.2 ✓ forcats 0.5.1## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## x dplyr::filter() masks stats::filter()
## x dplyr::lag() masks stats::lag()library(RSQLite)Download software and data
Download the data here.
sqlite <- dbDriver("SQLite")
DATA="/file/path/to/box/download"
WORK=DATADownload PRSice from https://choishingwan.github.io/PRSice/
wget https://github.com/choishingwan/PRSice/releases/download/2.3.3/PRSice_mac.zip- move the executable to the directory where you keep your software (optional)
** the first time you run PRSice, it will install additional components **
Terminal Setup
DATA="/file/path/to/box/download"
WORK=$DATA
PRSice=file/path/to/PRSice
cd $DATAExploration of the Data
dbname <- paste0(DATA,"repgp-data.sqlite3") ##This is just to create the file path to the sqlite3 file
## connect to db
db = dbConnect(sqlite, dbname)
## list tables
dbListTables(db)## [1] "ancestry_jkp" "ancestry_pop" "ancestry_supop" "users"dbListFields(db,"users")## [1] "id" "sample" "height" "weight" "human_id" "file_id" "blood"
## [8] "gender" "race"query <- function(...) dbGetQuery(db, ...)
users = query("select * from users")
names(users)## [1] "id" "sample" "height" "weight" "human_id" "file_id" "blood"
## [8] "gender" "race"Looking at distribution by count for each column
users %>% count(race)## race n
## 1 * 68
## 2 American Indian or Alaska Native 2
## 3 Asian 6
## 4 Black or African American 1
## 5 Caucasian (White) 1
## 6 Hispanic or Latino 2
## 7 Hispanic/Latino 3
## 8 White 167users %>% count(gender)## gender n
## 1 * 69
## 2 Female 46
## 3 Male 135users %>% count(blood)## blood n
## 1 * 111
## 2 A- 10
## 3 A+ 41
## 4 AB- 1
## 5 AB+ 4
## 6 B- 4
## 7 B+ 16
## 8 O- 11
## 9 O+ 52Plotting height distribution by gender
users %>% ggplot(aes(height,fill=gender)) + geom_density(alpha=0.6) + ggtitle("Height by gender - Missing gender, *, has bi-modal distr.")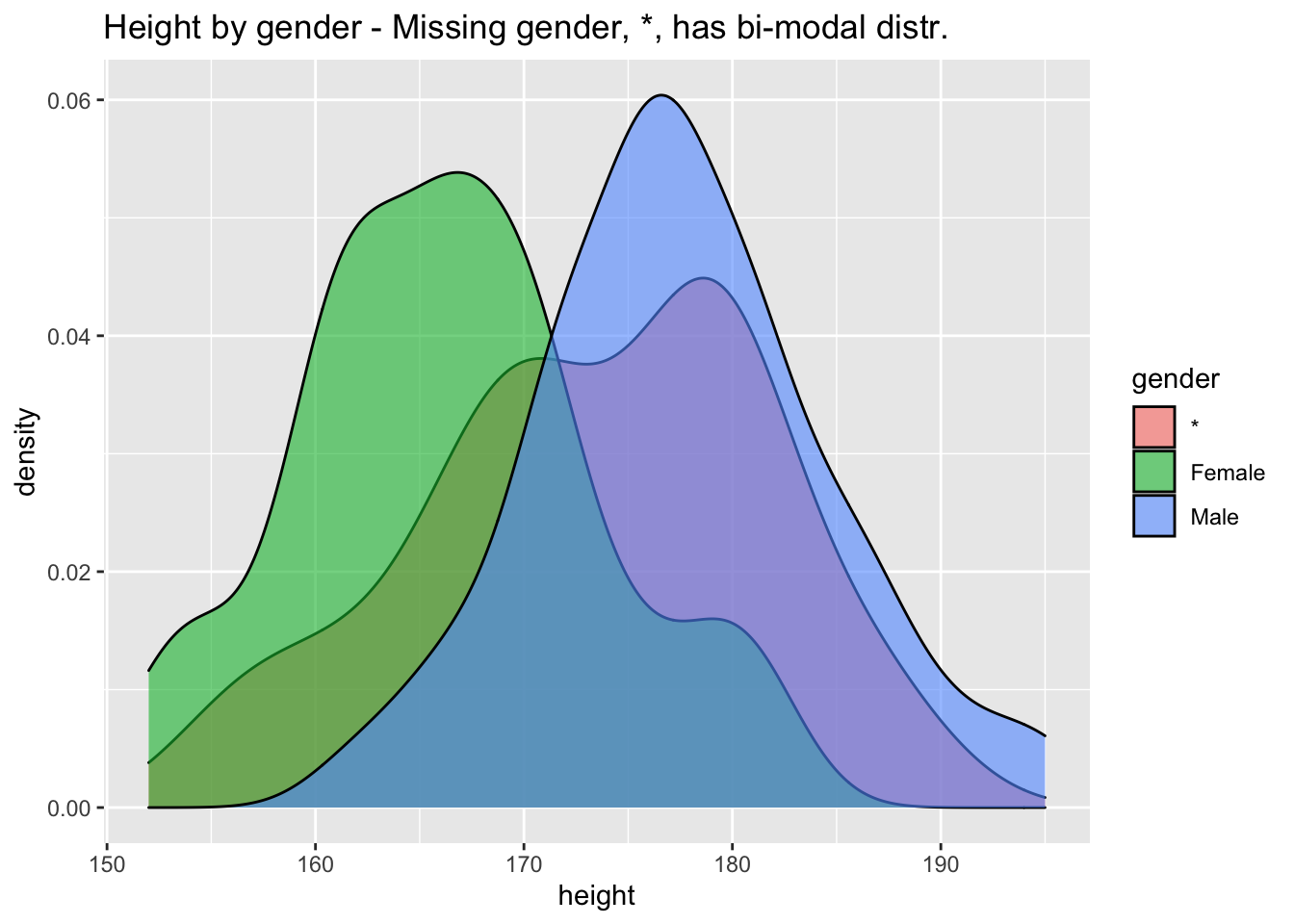
Running PRSice
fam = read_tsv(glue::glue("{DATA}/repgp.fam"),col_names = FALSE)## Rows: 568 Columns: 6
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (1): X2
## dbl (5): X1, X3, X4, X5, X6
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.names(fam)[1:2] = c("FID","IID")
fam <- fam %>% select(FID, IID) %>% inner_join(users %>% select(sample,height,weight,gender), by=c("IID"="sample"))
write_tsv(fam,file=glue::glue("{DATA}/phenodata.txt"))
mkdir $WORK/output
Rscript $PRSice/PRSice.R --dir $PRSice \
--prsice $PRSice/PRSice_mac \
--base $WORK/ukb_height.gz \
--target $WORK/repgp.chr# \
--snp variant_id \
--A1 effect_allele \
--A2 non_effect_allele \
--stat effect_size \
--beta \
--pvalue pvalue \
--pheno-file $WORK/phenodata.txt \
--pheno-col height \
--bar-levels 5e-08,5e-07,5e-06,5e-05,5e-04,5e-03,5e-02,5e-01,1 \
--fastscore \
--binary-target F \
--thread 2 \
--out $WORK/output/height_score_all
Plotting Observed Versus Predicted Height
## we already have the phenotype data in the fam variable
predicted_height = read_delim(glue::glue("{DATA}/output/height_score_all.best"),delim=" ")## Rows: 568 Columns: 4
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: " "
## chr (2): IID, In_Regression
## dbl (2): FID, PRS
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.combined <- predicted_height %>% inner_join(fam,by=c("IID"="IID"))
combined %>% ggplot(aes(PRS,height)) + geom_point()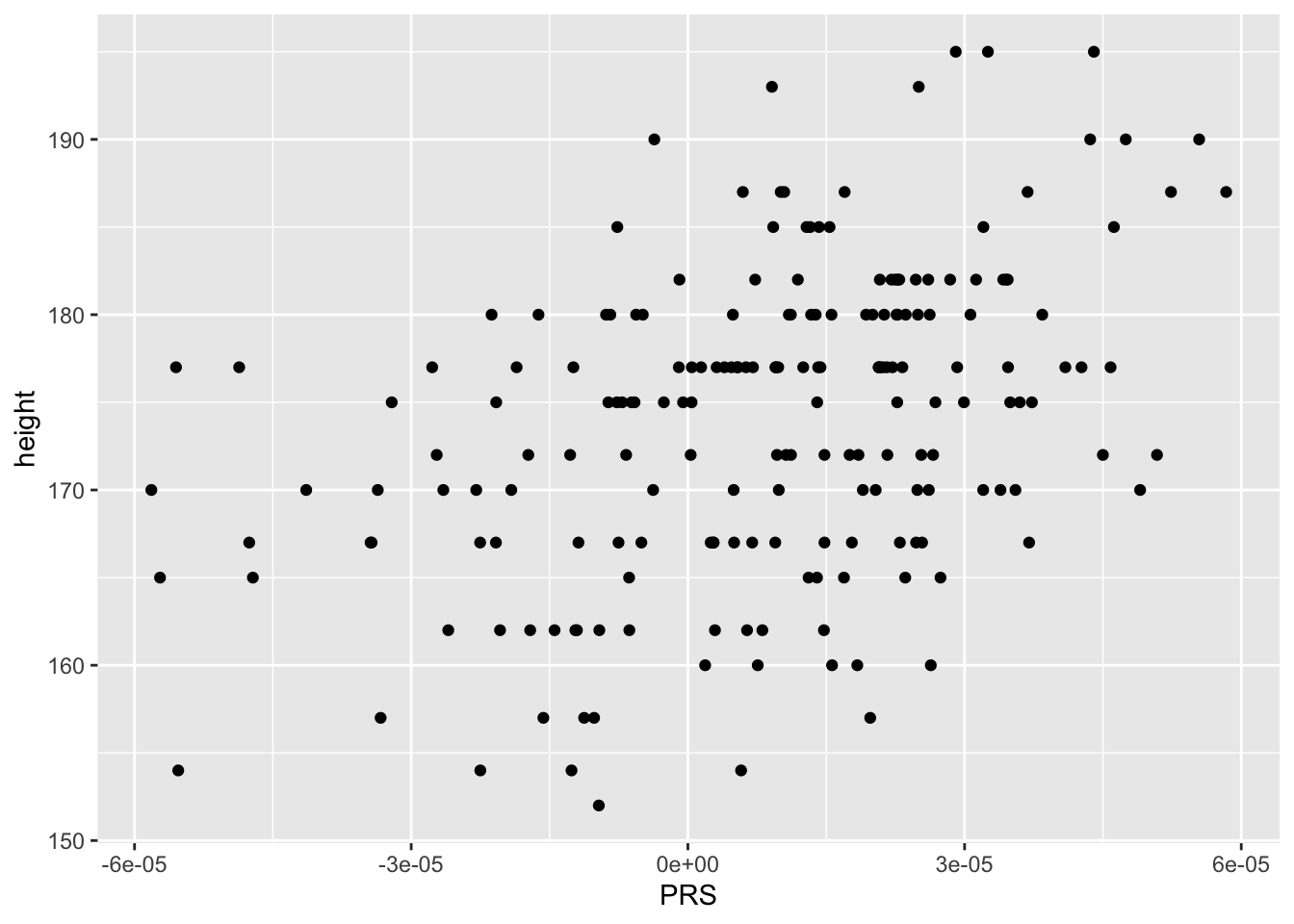
summary(lm(height ~ PRS,data=combined)) %>% coef() ## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 172.6716 6.075067e-01 284.230021 1.916175e-259
## PRS 159197.1399 2.478220e+04 6.423849 9.758137e-10