Learning Objectives
- Explain various methods to control for population structure
- Use mixed effects model approach to correct for population structure
- Use LD Score regression to distinguish between population structure and polygenicity driven inflation
Notes
Find the class notes here
Review connection between Zscore, p-value, Chi2 statistic
Notice that, under the null hypothesis, the estimated effect size divided by the standard error behaves like a normal(0,1) random variable when the sample size is large enough:
\[ Z = \frac{\hat\beta}{\text{se}(\hat\beta)}\] \[Z \approx N(0,1) ~~~~~~ \text{as } n \rightarrow \infty\] Let’s simulate zscore vector under the null hypothesis.
nsim = 5000
set.seed(2021021001)
zvec = rnorm(5000, mean=0, sd=1)Calculate the p-value (probability that a normal r.v. will be as large or larger in magnitude than the observed |zscore|
pvec = pnorm(-abs(zvec)) * 2 ## two-tailed
## check pvec is uniformly distributed
hist(pvec,main="is this uniformly distributed?")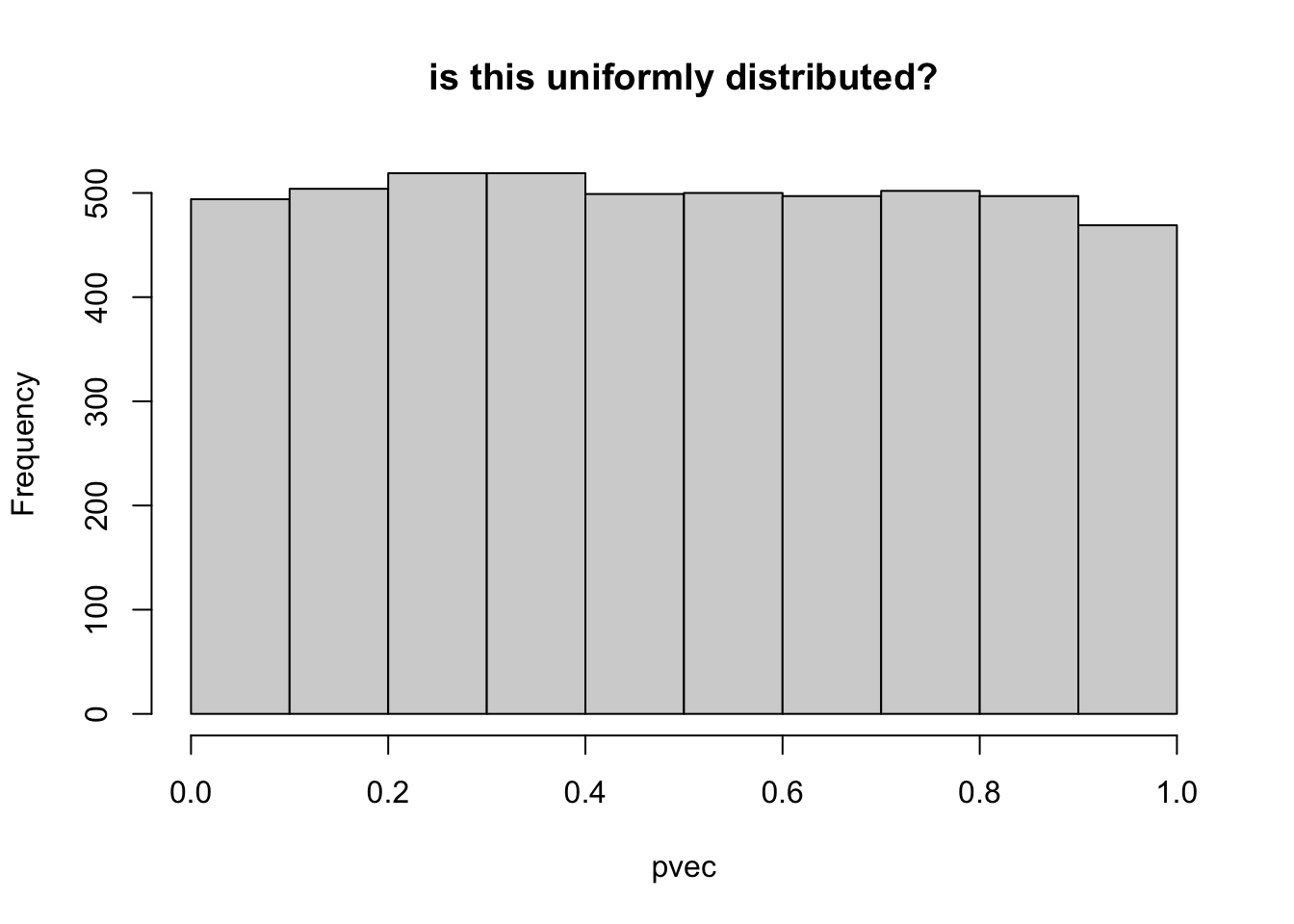
remember that if square the a normal r.v. you get chi2 r.v. with one degree of freedom
z2vec = zvec^2
## compare with chi2 rv. with 1 degree of freedom by simulating chi2,1 and qqplot
chi2vec = rchisq(nsim,df=1)
qqplot(chi2vec,z2vec,main="compare of zscore^2 and a chi2 r.v."); abline(0,1)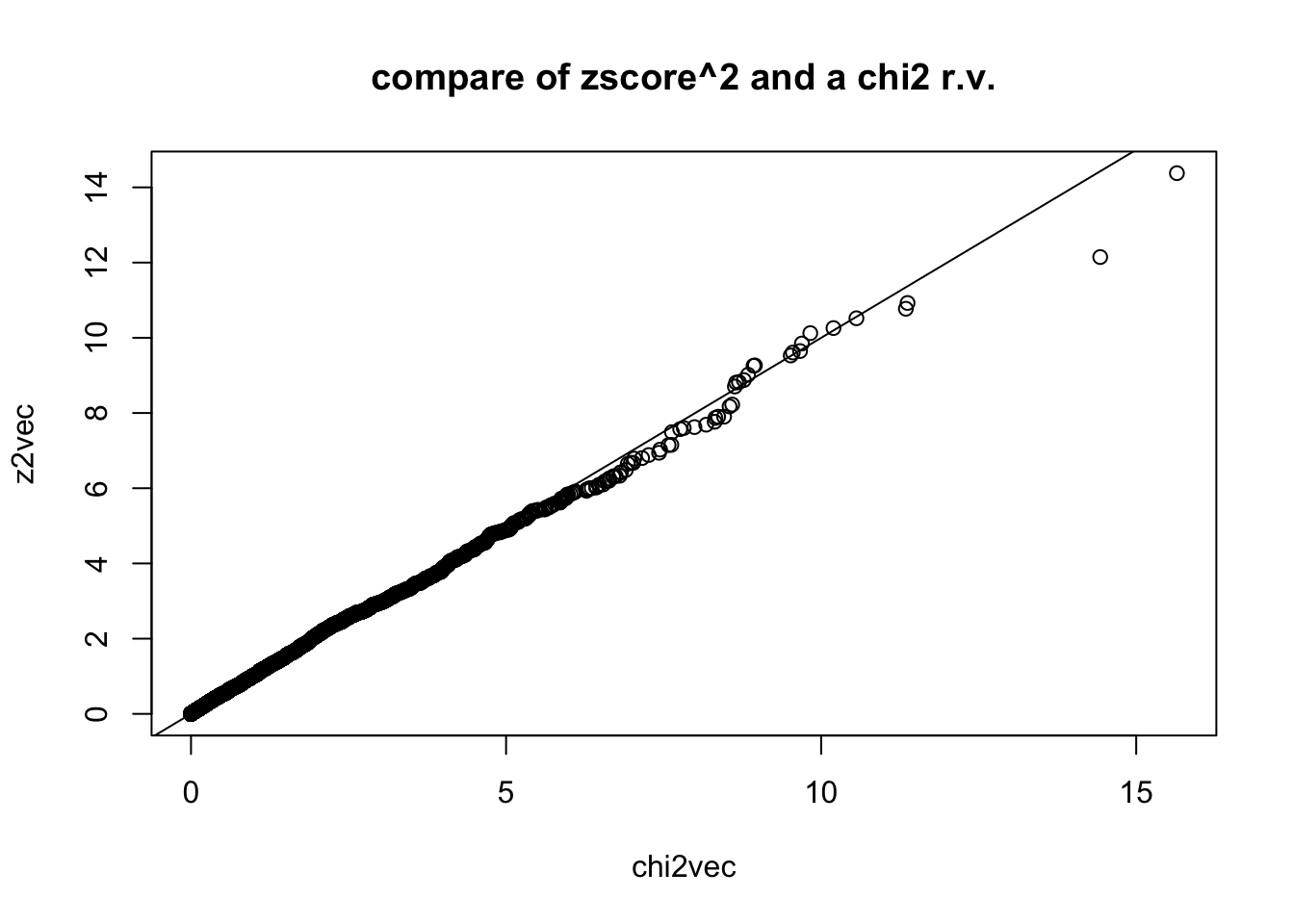
## for reference, let's compare two chi2,1 r.v.'s qqplot
chi2vec_2 = rchisq(nsim,df=1)
qqplot(chi2vec,chi2vec_2,main="qqplot of two chi2,1 r.v."); abline(0,1)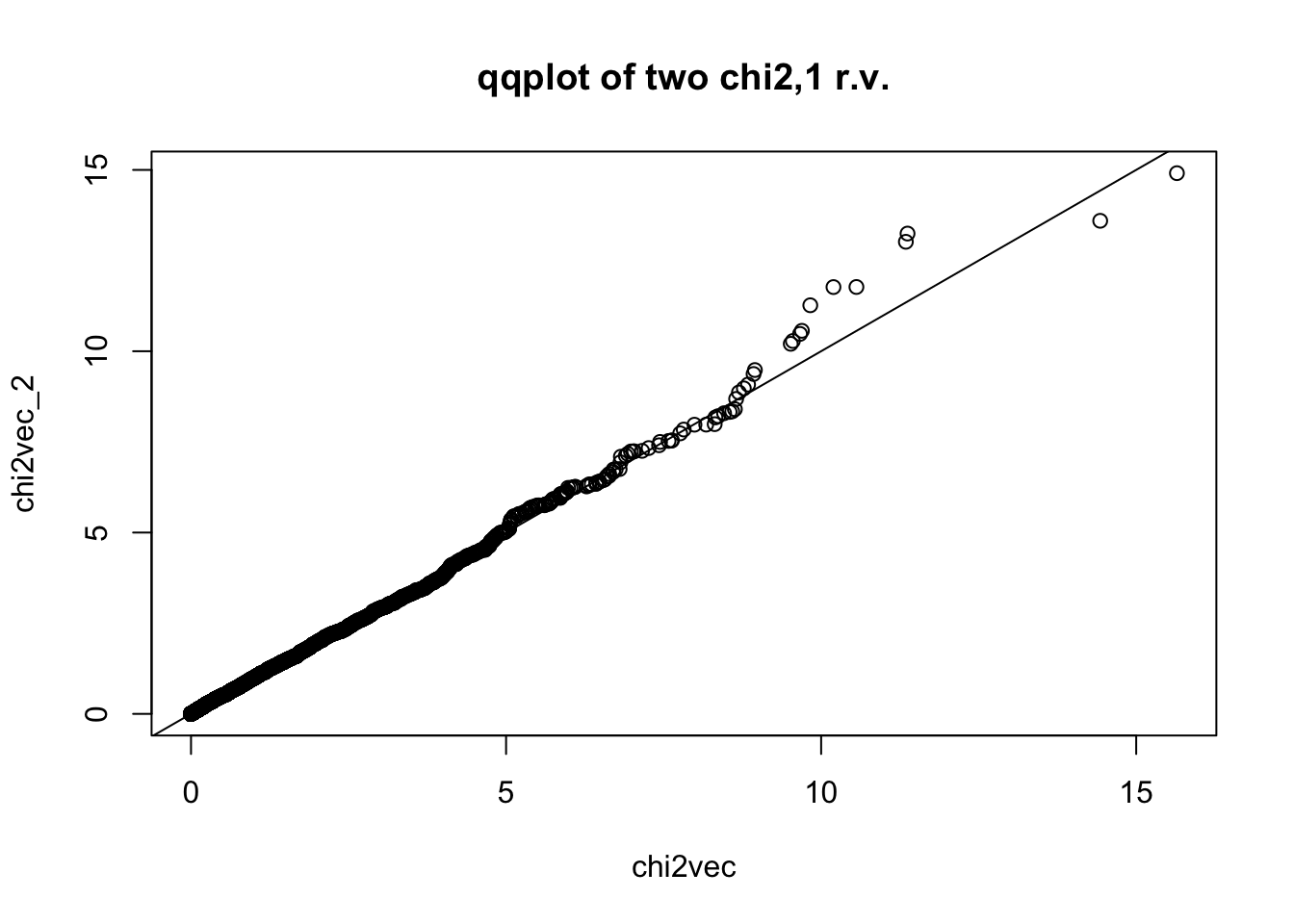
Sometimes you get the p-value instead of the zscore, you can generate chi2 by inverting the relationship.
chi2vec_from_p = qnorm(pvec / 2)^2
qqplot(chi2vec,chi2vec_from_p,main="chi2 from pvalue vs chi2 r.v."); abline(0,1)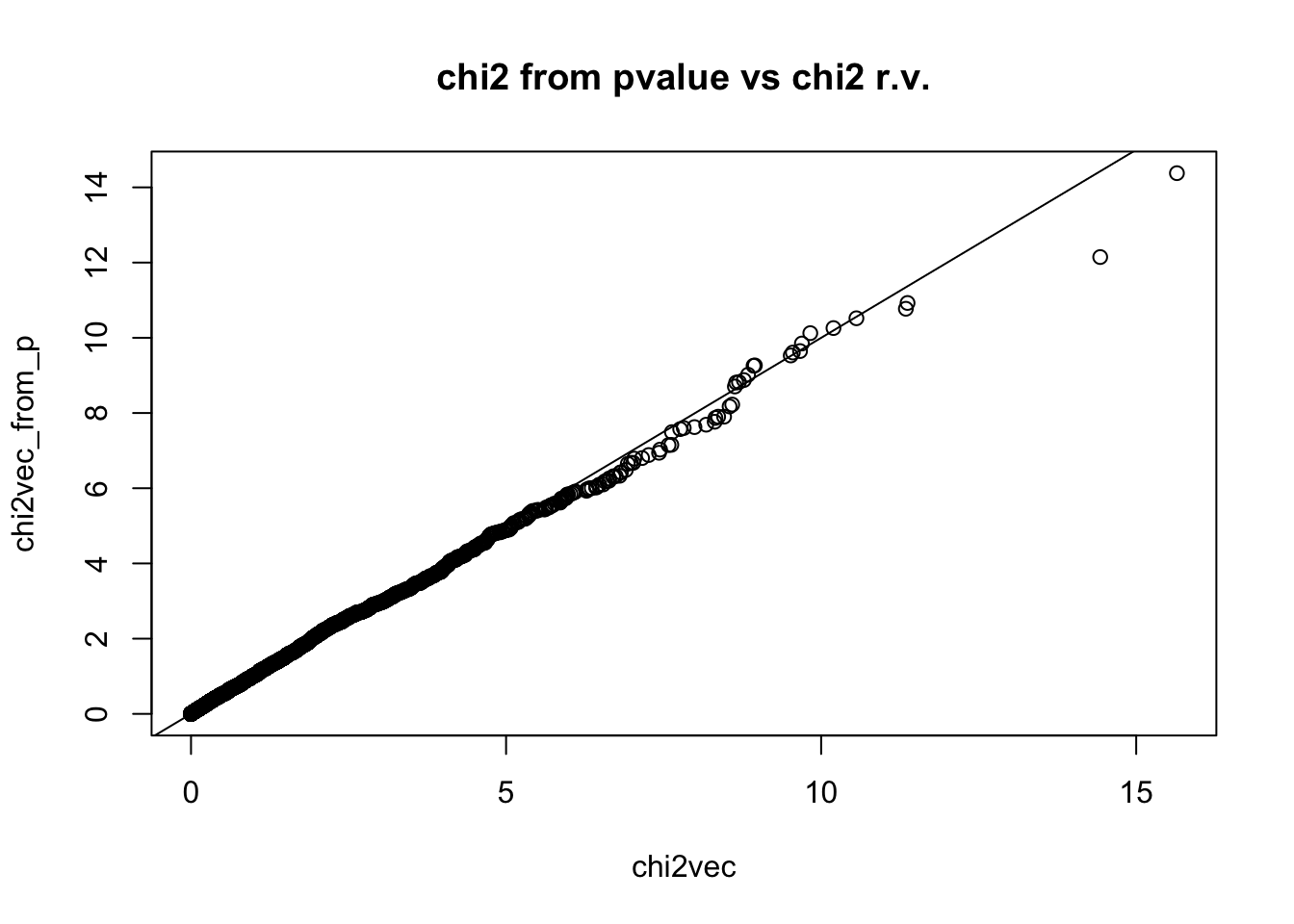
Homework problem
Write an R function to calculate the chi2 statistics as a function of the estimated effect size, \(\hat\beta\), and the standard error of the estimated effect size, se(\(\hat\beta\)).
References
- B. Devlin and Kathryn Roeder (1999) “Genomic Control for Association Studies”, Biometrics, Vol. 55, No. 4, 997-1004.
- H. M. Kang, J. H. Sul, S. K. Service, N. A. Zaitlen, S.-Y. Kong, N. B. Freimer, C. Sabatti, and E. Eskin, “Variance component model to account for sample structure in genome-wide association studies,” Mar. 2010.
- A. L. Price, N. A. Zaitlen, D. Reich, and N. Patterson, “New approaches to population stratification in genome-wide association studies,” Nat Rev Genet, vol. 11, no. 7, pp. 459–463, Jun. 2010. B. K. Bulik-Sullivan, P.-R. Loh, H. K. Finucane, S. Ripke, J. Yang, N. Patterson, M. J. Daly, A. L. Price, and B. M. Neale, “LD Score regression distinguishes confounding from polygenicity in genome-wide association studies,” Nat Genet, vol. 47, no. 3, pp. 291–295, Feb. 2015.